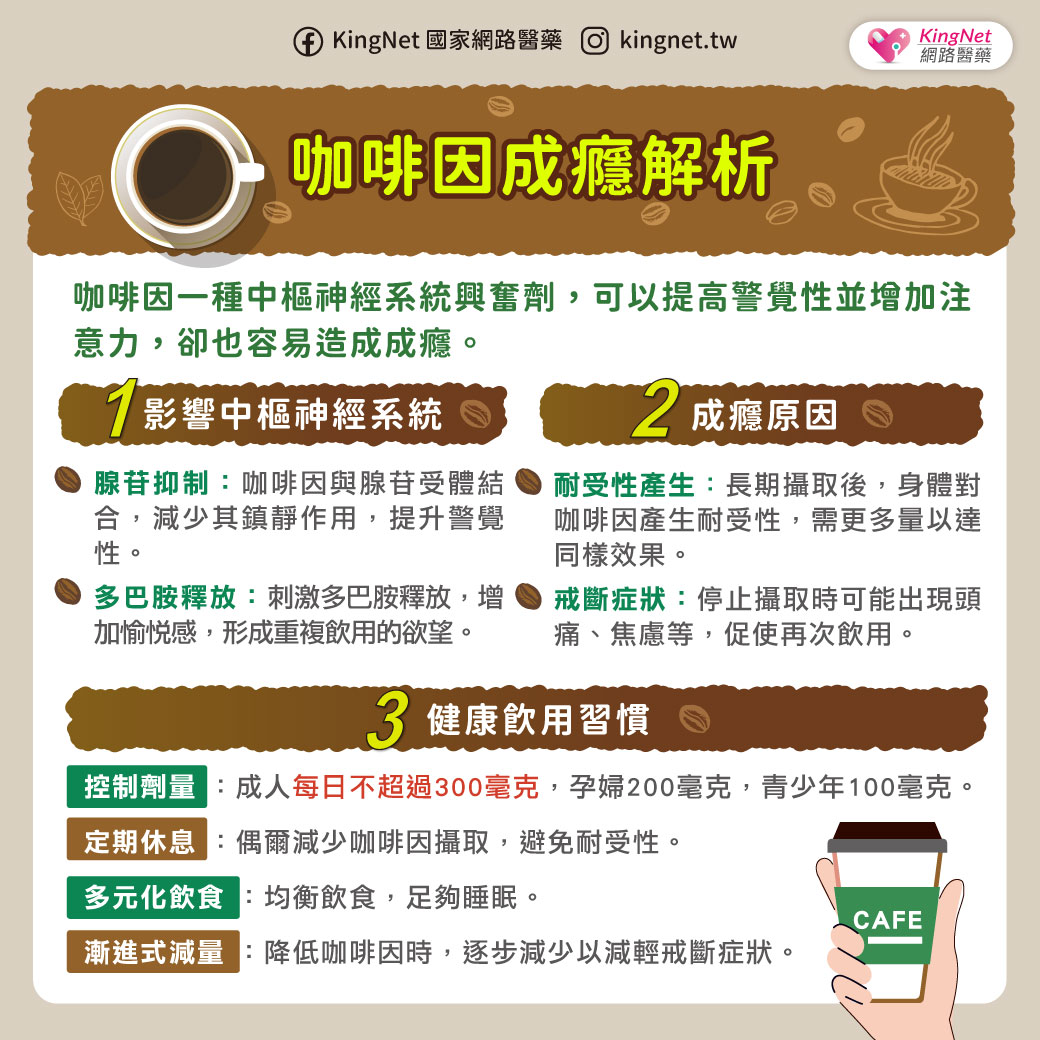
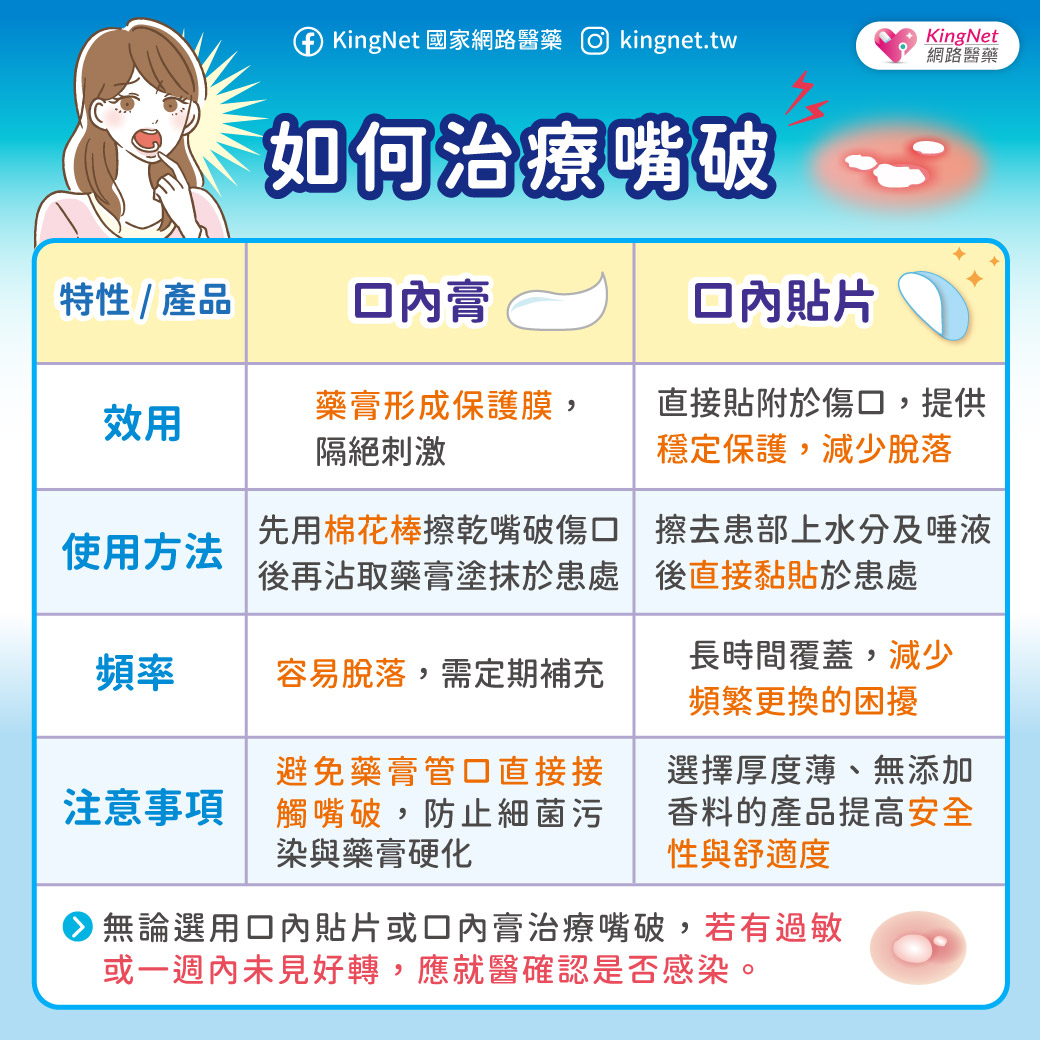
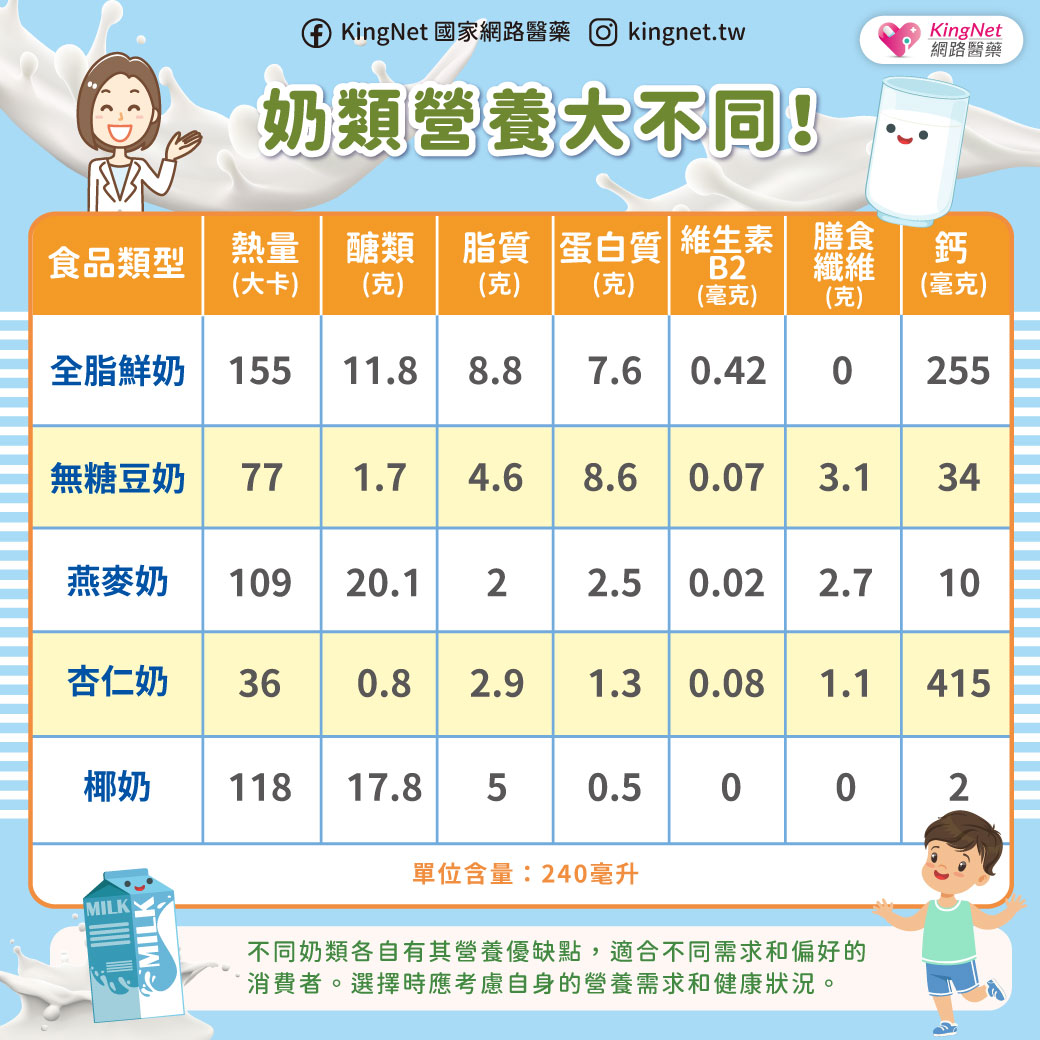
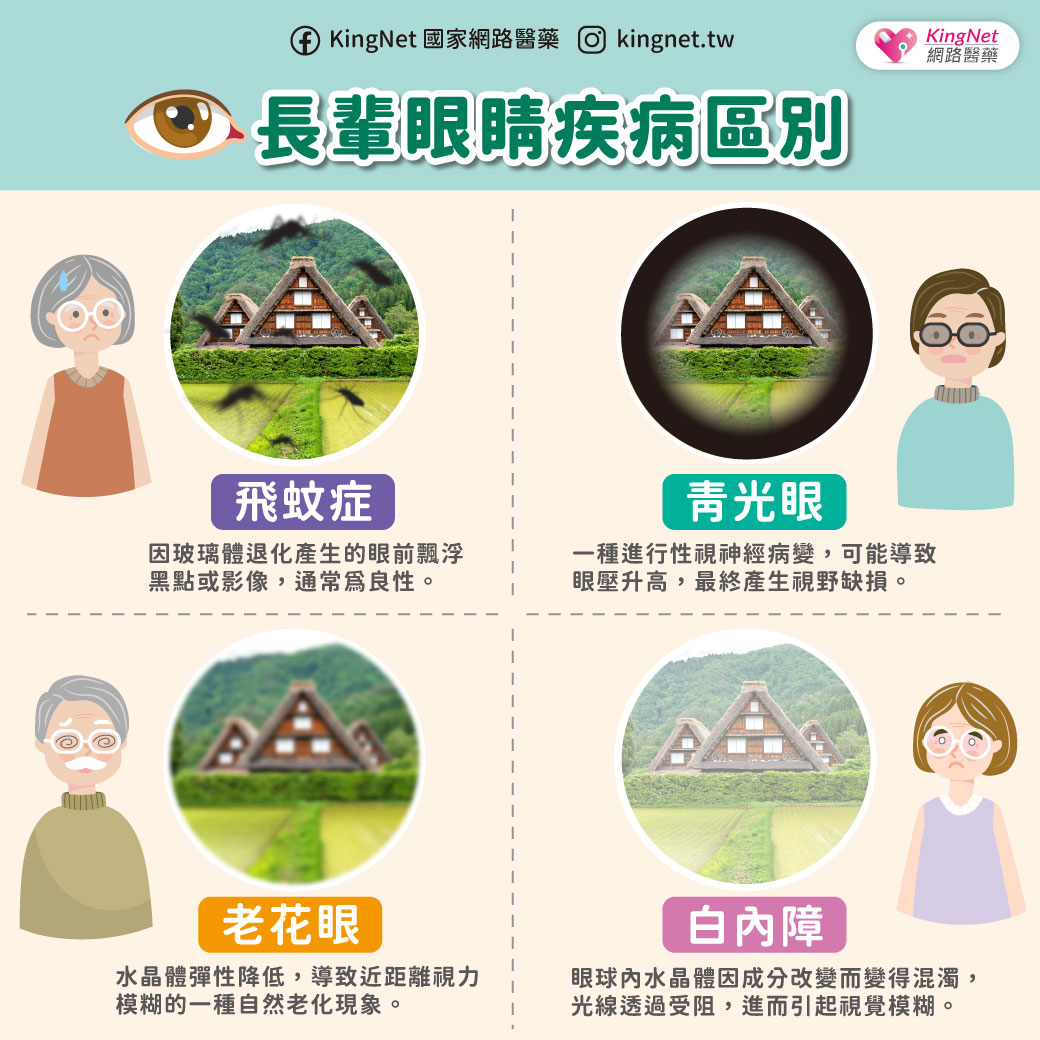
小腦萎縮症不是單一種疾病,而是一群症狀相似疾病的統稱,因為主要是脊髓及小腦功能失調的症狀,衛生署建議稱為「脊髓小腦共濟失調症」。發生的原因可能是脊髓病變,小腦不一定會萎縮。由於身體協調各肌肉收縮的功能不良,症狀為說話不清、吞嚥困難、寫字困難、手部細緻動作不良、走路不穩、顫抖、尿失禁等,多半在30至50歲之間開始出現症狀。脊髓小腦共濟失調症可分為遺傳型及散發型,遺傳性的患者會遺傳給下一代 ,發生率大約是每10萬人中有5~7人。診斷的方法有神經學檢查、神經系統磁振造影檢查、遺傳基因檢查。散發型共濟失調症的原因有酗酒、維生素B12缺乏、多發性硬化症、血管疾病、腫瘤等。
How to treat
目前醫學對本疾病仍無根治的方法,只能舒緩症狀及減緩惡化的進行,只有因為營養素缺乏、腫瘤所引起的共濟失調症狀,可以直接矯正這些原因。復健治療可改善一些症狀,使用手杖、助行器、輪椅可幫助患者行動,特殊輔具可幫助寫字、扣釦子、使用餐具等日常活動。語言治療可幫助改進說話的清晰度。
運動神經元疾病 (漸凍人)(Motor Neuron Disease)
運動神經元疾病的主要類型是肌萎縮性脊髓側索硬化症,由於運動神經元病變,使得神經無法控制肌肉所引起的問題。患者因為肌肉逐漸萎縮和無力,身體好像被逐漸凍結一樣,所以俗稱為“漸凍人”。肌肉由無力惡化至癱瘓,說話、吞咽和呼吸功能也減退,直至呼吸衰竭而死亡。運動神經元疾病並不影響患者的智力、記憶或感覺。發病的年齡多在40-60歲之間,發病後的存活生命大約在2-5年之間。發生率大約是10萬之5,10%的病例是遺傳性的,而其餘90%是散發性的。
How to treat
目前的治療方式或發展方向有神經保護劑、神經生長因子、幹細胞、基因治療等方面,現階段治療的效果還不理想。要安排患者房間環境的擺設、設計活動與運動、協助患者做運動以提升生活的品質。如果吞嚥功能下降,需攝取軟質、流質的食物。如果手的靈活度降低了,可用鬆緊帶或沾黏帶取代鈕釦、拉鍊,另外身體的清潔、排泄的照護、臥床者的按摩和翻身也是要注意的事項。
重症肌無力症(Myasthenia Gravis)
是一種自體免疫疾病,患者會產生異常的抗體,阻礙肌肉接受神經所釋放的乙醯膽素的訊息,所以肌肉收縮能力會下降。重症肌無力症的症狀是容易疲勞,肌肉會在短時間活動後便逐漸軟弱,眼皮下垂與看物品出現兩個影像常是最先出現的症狀,經過休息便能恢復,症狀惡化時出現吞嚥困難、言語不清、走路不穩等、脖子和肢體無力、呼吸的肌肉無力。重症肌無力較常發生在40歲以下的女性和50歲以上的人,發生率大約是萬分之1-2,患有其他自體免疫疾病的人罹患重肌無力症的機率較高,有5%的個案有遺傳的傾向。
How to treat
治療重症肌無力症的藥物有膽素酯酶抑制劑,使在神經肌肉交界處的乙醯膽素濃度提高,以彌補感受體的反應不足,另外還有免疫抑制劑、血漿分離術、免疫球蛋白等治療方法,如果發生呼吸肌肉痲痺,則需要呼吸器維持生命。經過治療,大多數患者可有正常的生活,有大約10%的重症肌無力病人有胸腺腫瘤,可考慮切除胸腺達到治療的目的。
How to treat
目前醫學對本疾病仍無根治的方法,只能舒緩症狀及減緩惡化的進行,只有因為營養素缺乏、腫瘤所引起的共濟失調症狀,可以直接矯正這些原因。復健治療可改善一些症狀,使用手杖、助行器、輪椅可幫助患者行動,特殊輔具可幫助寫字、扣釦子、使用餐具等日常活動。語言治療可幫助改進說話的清晰度。
運動神經元疾病 (漸凍人)(Motor Neuron Disease)
運動神經元疾病的主要類型是肌萎縮性脊髓側索硬化症,由於運動神經元病變,使得神經無法控制肌肉所引起的問題。患者因為肌肉逐漸萎縮和無力,身體好像被逐漸凍結一樣,所以俗稱為“漸凍人”。肌肉由無力惡化至癱瘓,說話、吞咽和呼吸功能也減退,直至呼吸衰竭而死亡。運動神經元疾病並不影響患者的智力、記憶或感覺。發病的年齡多在40-60歲之間,發病後的存活生命大約在2-5年之間。發生率大約是10萬之5,10%的病例是遺傳性的,而其餘90%是散發性的。
How to treat
目前的治療方式或發展方向有神經保護劑、神經生長因子、幹細胞、基因治療等方面,現階段治療的效果還不理想。要安排患者房間環境的擺設、設計活動與運動、協助患者做運動以提升生活的品質。如果吞嚥功能下降,需攝取軟質、流質的食物。如果手的靈活度降低了,可用鬆緊帶或沾黏帶取代鈕釦、拉鍊,另外身體的清潔、排泄的照護、臥床者的按摩和翻身也是要注意的事項。
重症肌無力症(Myasthenia Gravis)
是一種自體免疫疾病,患者會產生異常的抗體,阻礙肌肉接受神經所釋放的乙醯膽素的訊息,所以肌肉收縮能力會下降。重症肌無力症的症狀是容易疲勞,肌肉會在短時間活動後便逐漸軟弱,眼皮下垂與看物品出現兩個影像常是最先出現的症狀,經過休息便能恢復,症狀惡化時出現吞嚥困難、言語不清、走路不穩等、脖子和肢體無力、呼吸的肌肉無力。重症肌無力較常發生在40歲以下的女性和50歲以上的人,發生率大約是萬分之1-2,患有其他自體免疫疾病的人罹患重肌無力症的機率較高,有5%的個案有遺傳的傾向。
How to treat
治療重症肌無力症的藥物有膽素酯酶抑制劑,使在神經肌肉交界處的乙醯膽素濃度提高,以彌補感受體的反應不足,另外還有免疫抑制劑、血漿分離術、免疫球蛋白等治療方法,如果發生呼吸肌肉痲痺,則需要呼吸器維持生命。經過治療,大多數患者可有正常的生活,有大約10%的重症肌無力病人有胸腺腫瘤,可考慮切除胸腺達到治療的目的。